






色彩靚麗 工藝多樣

24小時(shí)客服熱線
COMPANY PROFILE
臨沂貝殼屋新材料科技有限公司,是一家專(zhuān)門(mén)從事貝殼粉涂料、藝術(shù)涂料、無(wú)機(jī)涂料等環(huán)保涂料研發(fā)、生產(chǎn)及銷(xiāo)售的股份制公司,公司團(tuán)隊(duì)由國(guó)內(nèi)從事貝殼粉等環(huán)保涂料研究和生產(chǎn)的專(zhuān)家組成。
PRODUCT & APPLICATION EFFECT
CONSULTING TRENDS
PRODUCT ADVANTAGE
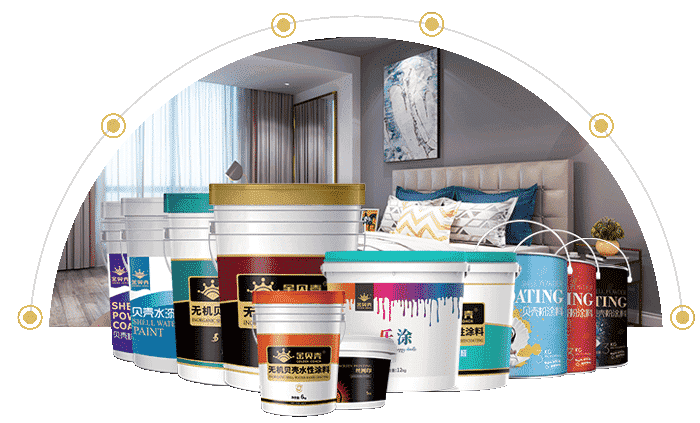
Green Health
Beautiful colors
Soft touch
Diverse craftsmanship
Visual softness
Friction resistance
友情鏈接: 展臺(tái)搭建制作整木定制十大建材品牌蚌埠裝修成都裝修公司櫥柜加盟衣柜十大品牌全屋定制著名品牌水泥路面修補(bǔ)家居定制全屋定制環(huán)氧地坪漆裝修公司







































 查看全國(guó)代理
查看全國(guó)代理